CSHL + Arizona visit
We had a super busy trip to America! Aparna and Eyal represented the lab in the Cold Spring Harbor Lab meeting on Biology and Genomics

We had a super busy trip to America! Aparna and Eyal represented the lab in the Cold Spring Harbor Lab meeting on Biology and Genomics

Max has some preliminary results from training a machine learning model (deep network) to identify and track ants in videos with multiple ants. You can

After considerable delays…. Hive2, the new computer cluster for our Faculty, finally arrived on campus today. It was assembled by Matrix at their facility, now

Now this event was truly something else! We travelled all the way down to Avdat in the Negev highlands to celebrate the planting of a

We had two PhD proposal presentations in the departmental student forum today! Yoann presented his project titled: “Exploiting the evolution of odorant receptors in ants

Pnina’s paper on ancient DNA population genomic analysis of grapes from the Byzantine era is out in PNAS!https://www.pnas.org/doi/10.1073/pnas.2213563120 Ancient DNA is so cool! It allows

The 4th meeting of the Israeli Society of Evolutionary Biology was hosted by the Institute of Evolution, co-organized by Eyal Privman and Eran Tauber. About

Our 8th Haifa Winter Advanced Bioinformatics Workshop attracted a diverse assemblage of students and researchers, and included this time a number of lecturers from abroad

Yoann, Felix, Volans and Eyal attended Camp Evolution VIII in Sede Boqer. This is a unique one-week course organized by Ariel Novoplansky. Every time Ariel
אתמול היה יום מוצלח במיוחד ליחסי הציבור של הנמלים! איל דיבר בתכנית העולם היום (כאן 11) על מחקר חדש שחישב כמה נמלים יש בעולם, ובתכנית שלושה


The lab got a new 5-year ISF grant to study the supergene we discovered in the desert ant Cataglyphis niger. Graduate student and postdoc positions are

Eyal is back from the one year sabbatical in London where he refined his expertise in Bayesian population genomics and the art of luggage packing.

Our lab participated in IUSSI 2022 (the meeting of the International Union for the Study of Social Insects) in San Diego, which is the ultimate
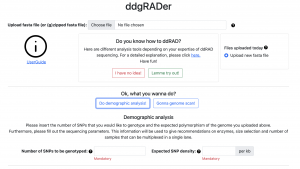
Just in time for the IUSSI meeting in San Diego, Felix completed the development of the web tool ddgRADer: http://ddgrader.haifa.ac.il/ This user-friendly tool will assist

Felix have just received, safe and sound, the shipment of a large collection of samples from Deborah Gordon’s lab. These priceless samples were collected over

Eyal started a one-year sabbatical in University College London, hosted by two hosts: Ziheng Yang and Seirian Sumner. The Yang lab develops methods in molecular

Our lab expanded our phylogenetic scope beyond the Cataglyphis bicolor species group and into the C. albicans sister clade. We went on a two day sampling trip

Mazal tov! We celebrated Pnina’s PhD today with a concluding PhD lecture (“public defense”) and the general ceremony of the University for awarding the PhD

Not yielding to the coronavirus! Eyal visited Jessica and Alan in Riverside, to discuss the BSF project on genome evolution (linkage mapping 10 formicine species),
Welcome Yoann, Felix and Volans! The lab expanded significantly in the last few months, despite substantial COVID challenges. Yoann was the first to make it
We concluded our 7th Haifa Winter Workshop in Bioinformatics, which was focused on multi-omics and machine learning applications in omics. It was especially challenging this
Aparna just finished constructing RADseq libraries for her project on social polymorphism in Cataglyphis niger, and shipped them for sequencing. She broke the lab record
Over the last days we participated in two Israeli meetings. These were online meetings, obviously, so unfortunately no photos. But it quite was an experience!
איל התראיין בתכנית שלושה שיודעים ברדיו כאן תרבות לגבי מחקר חדש שמתאר איך השרירים של הנמלים התאימו את עצמם באבולוציה לסחיבת משאות כבדים אחרי אובדן
Two upcoming Israeli meetings are now open for registration and abstract submission: Israeli Society for Evolutionary Biology Israel Zoological Society (Eyal is involved in the

Doaa Tehawey and Yara Ghnamah concluded their third-year projects and presented them today as posters! They both managed to finish and wrap up their analyses
איל התראיין אצל דודו ארז בתכנית שלושה שיודעים (רדיו כאן תרבות) בנושא נמלת גיהנום שנמצאה כלואה בענבר בן 100 מיליון שנה כשבפיה לכוד טרף (תיקן
Our lab received another NSF-BSF grant(!!!) in collaboration with the labs of Mickey Kosloff (at Haifa) and Juergen Liebig (at ASU). We are recruiting Ph.D.

Today we (Eyal and Aparna) went out sampling Cataglyphis for the first time of this reproductive season, looking for drones (for their haploid genomes). We
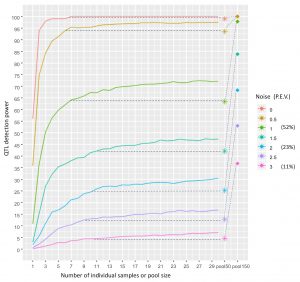
Shani and Pnina’s paper was published in PLoS Computational Biology! https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007653 This methodological study was actually a pilot for our GWAS of cuticular hydrocarbons. We
במסגרת סדרת ההרצאות לקהל הרחב ״מדע מההר״ המיוחדת לתקופת הקורונה, איל נתן את ההרצאה הראשונה (ולכן סלחו בבקשה על הבעיות הטכניות הקלות) בנושא ״פרדוקס האבולוציה
Our lab received an NSF-BSF grant in collaboration with the lab of Deborah Gordon at Stanford! We have advertised Ph.D. student and postdoc positions: NSF-BSF
״עושים היסטוריה עם רן לוי״ עשו פודקאסט נהדר על ״מה אפשר ללמוד מנמלים״ שבו גם התארח איל https://www.ranlevi.com/2020/01/20/ep299-ants/

Aparna and Doaa are conducing 1000 aggression assays to determine which of the 30 nests we sampled are separate monogyne colonies (so they aggress ants
Our lab participated in the meeting of the Israel Zoological Society (in Jerusalem) and the first ever meeting of the Israeli Society of Evolutionary Biology
We finally have the results from our genomic mapping of cuticular hydrocarbons (CHC) in Cataglyphis niger, which are known to be the chemical cues used
איל סיפר על הנמלים שחיו שנים בבונקר גרעיני נטוש עד שבאו חוקרים פולנים והצילו אותן, וגם חשפו את סודן האפל. ראיון בתוכנית שלושה שיודעים ברדיו
Our paper is online at Ecology and Evolution! This study was the Ph.D. project of Viet-Dai Dang, at the lab of John Wang. Dai carried
Eyal is busy nowadays writing the next ISF grant for continued genomic work in the Cataglyphis social polymorphism system, and the lab is very busy

This year, Eyal and Pnina represented the lab in SMBE and ESEB. Pnina presented a poster on her mutation rate study and Eyal gave a

Our paper is online at Scientific Reports! https://www.nature.com/articles/s41598-019-45950-1 This was a collaboration led by Tali Reiner Brodetzki and Abraham Hefetz (TAU) to resolve species delimitation
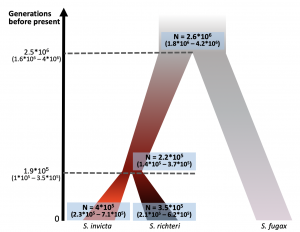
Pnina’s paper was published in BMC Evolutionary Biology! https://bmcevolbiol.biomedcentral.com/articles/10.1186/s12862-019-1437-9 This is a demographic history study that developed from our collaboration with DeWayne Shoemaker. Pnina used
Tal’s paper was published in Scientific Reports! https://www.nature.com/articles/s41598-019-42795-6 We evaluated the benefit of using a haploid sample in a denovo genome sequencing project. We saw

On the first really hot day of spring Camponotus have their mating flights. Today we collected C. fellah from the population that lives on campus

We kick started our joint BSF project on genome evolution in ants. Jessica Purcell and Alan Brelsford visited us in Haifa and we started to

איל נתן הרצאה על ״פרדוקס האבולוציה של אלטרואיזם״ במועדון של קיבוץ מעברות

Our workshop on “Omics for environmental and host-associated microbiomes” was a smashing success (see the program). With more than 40 participants we could not find
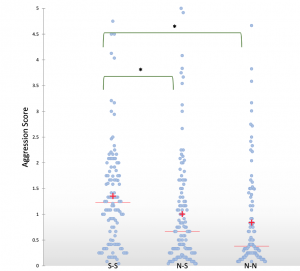
Shani found an interesting link between the level of aggression between workers from different monogyne Cataglyphis niger colonies and whether or not sexuals (gynes or

איל ידבר על ״פרדוקס האבולוציה של אלטרואיזם״ במסגרת סדרת ההרצאות ״מדע על ההר״, המיועדות לקהל הרחב
We recently sent for publication the first draft genome for Cataglyphis niger, which was done by Tal using high-coverage (>100X) Illumina sequencing of a single
התפרסם מחקר מהמעבדה של לורן קלר בשוויץ שהראה שנמלים שנדבקות במחלה מרחיקות את עצמן משאר חברותיהן, ובכך מצמצמות את הסיכון להתפשטות המחלה בקן. הארץ פרסם
עוד ראיון אצל דודו ארז בתכנית שלושה שיודעים ברדיו כאן תרבות על טרמיטים ביפן שאיבדו את המין הזכרי – הקלטה בלינק
Amir’s study of the odorant receptors (ORs) on the fire ant social chromosome was published in Genome Biology and Evolution: https://academic.oup.com/gbe/advance-article/doi/10.1093/gbe/evy204/5100826 We discovered a cluster

This year we had the most important meeting for ant researchers – the international conference of the IUSSI that takes place only once every four
בעבקבות הכתבה בהארץ איל התראיין בתכניות רדיו על האבולוציה של נמלי האש ובכלל על סיפורי נמלים: ברדיו קול רגע אצל חיים הכט וברדיו כאן תרבות

We organized a German-Israeli Minerva School on “New Frontiers in Sociobiology and Sociogenomics” together with Guy Bloch from the Hebrew University, Juergen Gadau from the

התפרסמה בהארץ כתבה על מחקר שלנו: נמלי האש משגשגות בזכות כישורים חברתיים
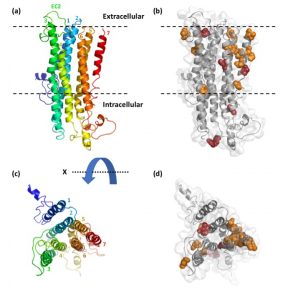
Rana’s evolutionary study of the gene family of the odorant receptors (ORs) in ants was published in Genome Biology and Evolution! We found evidence for

Our new Tecan liquid handling robot is here! It started to learn the TruSeq protocol for constructing genomic libraries. Shani will use it to construct
We won a BSF grant together with Jessica Purcell and Alan Brelsford from UC Riverside, and also in collaboration with the GAGA consortium. In this
איל התרייאן בלונדון את קירשנבאום על מחקר שהראה שנמלים אפריקאיות שמתמחות בצייד טרמיטים מפנות את הפצועות משדה הקרב ומעניקות להן טיפול מציל חיים https://www.youtube.com/watch?v=LFHX0AYeBns

Rana and Pnina received their M.Sc. diplomas! Pnina is already on track for her direct Ph.D. and Rana will hopefully also come back soon to
A big day for the lab! We got the sequencing results for the first RADseq (Restriction-site Associated DNA sequencing) library, and they look great! Shani
Gratulations Tal on the best poster prize in the meeting of the Zoological Society of Israel!
We attended two insect/social-insect conferences this summer: the meeting of the European Sections of the IUSSI in Helsinki and the American IUSSI satellite meeting of

Eyal gave a talk in the Nola Socks pub on “The paradox of the evolution of altruism”, from Darwin’s ants to our genomic age research

איל יתן מחר הרצאת מדע-על-הבר בנושא ״פרדוקס האבולוציה של אלטרואיזם״ בפאב נולה סוקס

This was it! after several failed attempts we came back to the Dead Sea with full intent not to let those difficult Camponotus cericeus ants

IBS 2016 (http://ibs2016.haifa.ac.il/) For the first time in IBS history we hosted the symposium at the University of Haifa, and it was a huge success!

We (Daniel Sher and Eyal Privman) held a whole week workshop on transcriptomics at the University of Haifa. Five full days packed with lectures and
Together with Daniel Sher and additional invited speakers. February 21-25, 2016, 8:30-16:30. More details in this Advertisment Booked out

First footage of beautiful tandem runs of Camponotus sericeus ants in our lab. These ants were collected south of the Dead Sea to participate in

We celebrated moving into our new lab (the construction was actually completed in September). We had a great crowd of friends and colleagues gathering together

We celebrated the signing of the ATP (for those unfamiliar with hi-tech jargon: Acceptance Test Procedure) of our Faculty’s new computer cluster: The Hive. We
Our lab received generous funding from the ISF that will allow a new research program into the evolution and genomic basis of social recognition in

In addition to the BSF funding of our collaboration with DeWayne Shoemaker’s lab at the USDA, we now received the Bergmann Award: http://www.bsf.org.il/bsfpublic/DefaultPage1.aspx?PageId=697&innerTextID=697

Our lab starred in IBS 2015! Rana presented the first poster from our group on the evolution of odorant receptors in seven ant species and

We began our Cataglyphis digging season in Bezet beach (close to Rosh Hanikra). It was lots of fun and hard work! We came back with
מכתב עליו חתומים חוקרים ישראלים בתחומי האבולוציה והאקולוגיה מכל האוניברסיטאות התפרסם ביום שישי (9/1/15) במוסף ספרים של “הארץ”: http://www.haaretz.co.il/literature/letters-to-editor/.premium-1.2529953 המכתב שלנו הוא תגובה למאמר
A new grant from the U.S.-Israel Binational Science Foundation (BSF) will fund research in collaboration with Dr. DeWayne Shoemaker (USDA) on the population genomics in
The existing M.Sc. program in bioinformatics will be broadened next year. The program is currently within the Department of Evolutionary and Environmental Biology. Next year
The lab can now hire a researcher or postdoc from a recently won grant (Marie Curie CIG), which may develop to a permanent position. Candidates
Graduate student positions are available! If you are interested in computational biology research and the evolutionary genomics of social insects please to contact us. Candidates should have